

|
||||||||
| Genomic Services | ||||||||
|
||||||||
| Protein Services | ||||||||
|
||||||||
Located in rooms
B065 and B017 |
| DNA Sequencing/Fragment Analysis |
• DATA ANALYSIS |
|
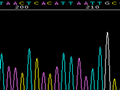
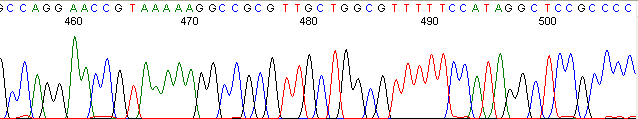
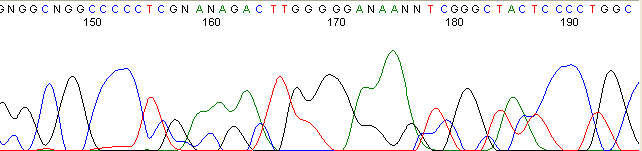
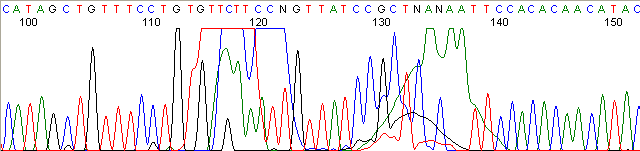
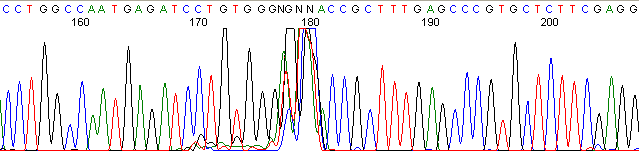
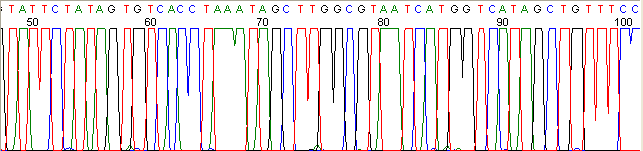
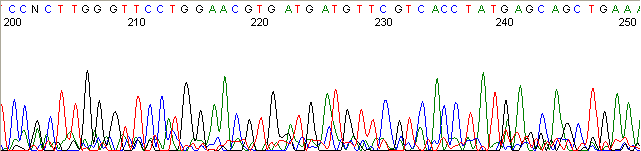
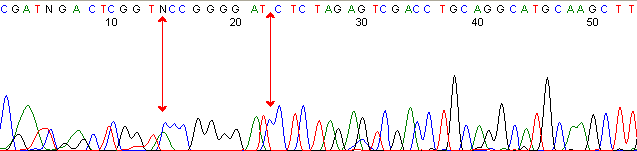
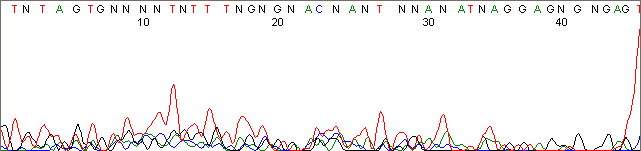
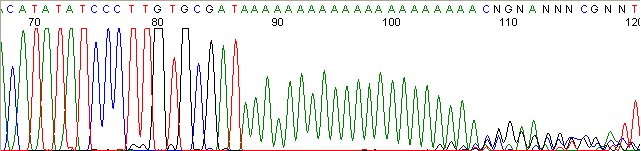
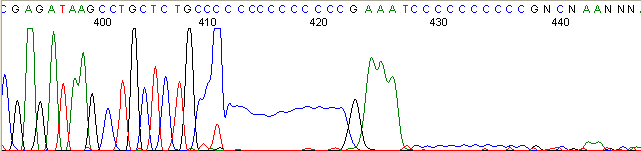
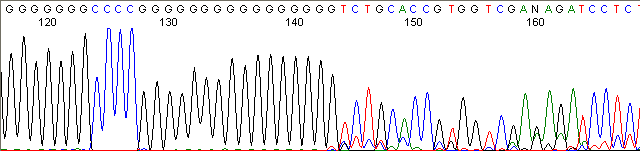
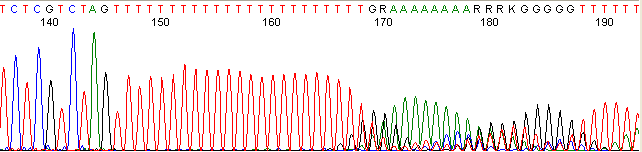
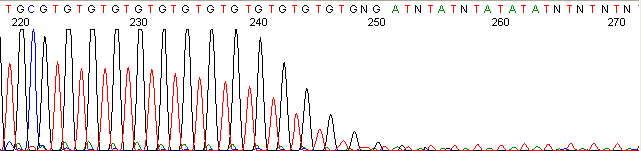
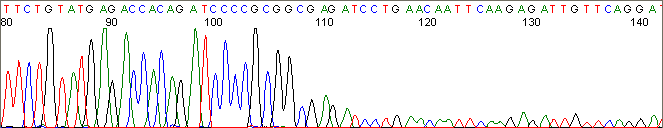
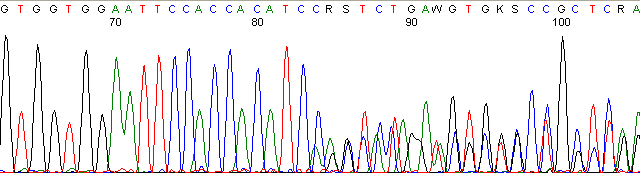
Below are graphical examples of common DNA sequencing issues. Most fair/poor sequencing data require simple adjustments to fix. The PAN Facility offers FREE reruns and/or resequencing of most fair and poor data. Bad data usually requires a little more troubleshooting. If you have any questions or concerns about your data please contact us at dnaseq@stanford.edu or 650-723-3189. **Due to changes in ABI polymer formulations, please be aware the first ~20 bases of a sequence may be unreadable due to poor resolution.
Poor Resolution (top)
Dye Blobs (top)
Dye Spikes (top)
Top Heavy (top)
Weak and Noisy Signal (top)
Missed Calls (top)
No Data (top)
Homopolymer Region (poly A, T, C, G) (top)
Repeat Region (top)
Secondary Structures / Hairpins (top)
Contaminated Sequence (top)
Helpful links (top) Applied Biosystems - DNA Sequencing by Capillary Electrophoresis Qiagen - The QIAGEN Guide to Template Purification and DNA Sequencing (2nd Edition) Roswell Park Cancer Institute DNA Sequencing |
Home | FAQs | | Prices | Contact Us | Publications | Feedback
Beckman Center | Stanford Medical Center | Stanford University
© 2006 Stanford PAN Facility. All rights reserved